Precision
in proteomics.
Fluorosequencing moves beyond genomics as the world's first single- molecule protein sequencer.
Ask more
precise questions.
Fluorosequencing goes where existing technologies cannot, empowering scientists to ask key questions that will help overcome difficult-to-treat diseases.

Transform the way we manage disease.



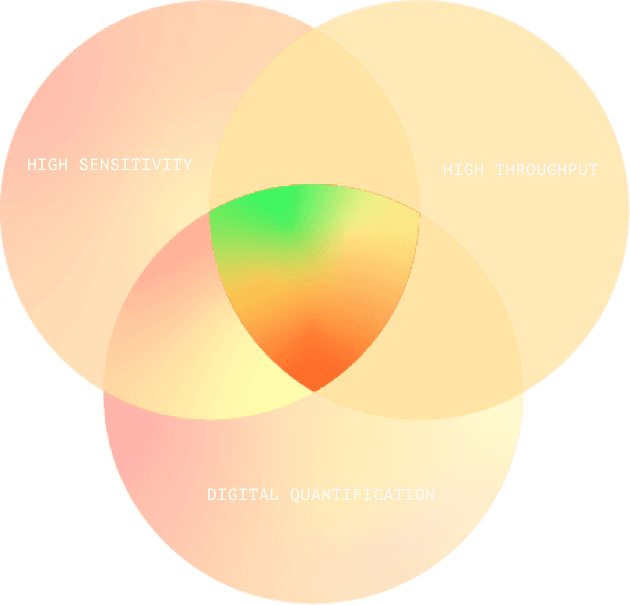
Fluorosequencing’s Sweet Spot
Fluorosequencing brings all the advantages of next- generation DNA sequencing to proteins, combining leading-edge digital quantification with high sensitivity and throughput. It is ideal for quantifying vast numbers of peptides in ultra-low sample concentrations, such as those found in tumor biopsies, or in extremely rare samples.
Furthermore, Mass Spectrometry is not suitable for many applications because of its low sensitivity and throughput.
1. Liu, Y., Beyer, A. & Aebersold, R.Cell165, 535–550(2016).
Fluorosequencing offers the ability to sequence hundreds of millions or billions of individual protein molecules simultaneously.
Competition
- Indirect measurement
- No PTM information
- Poorly correlated quantification
- High sample requirement
- Sequential processing
- PTM low resolution
- Qualitative measurement
- Biased identification
- Variable Specificity
- Ultimate sensitivity
- Ultra-low sample requirement
- Massively parallel throughput
- Absolute quantification
- De novo PTM discovery

SINGLE-MOLECULE SENSITIVITY
Fluorosequencing is 1 million times more sensitive than mass spectrometry. Assuming a factor of at least 1000, this translates into extremely low sample amounts: the difference between needing 1/4th of a pancreas or sampling with a needle for biopsy material.

MASSIVELY PARALLEL ARCHITECTURE
Our technology achieves a scalable throughput that was previously unattainable, empowering researchers and clinicians to identify hundreds of millions or even billions of peptide molecules on a single glass slide.

ABSOLUTE QUANTIFICATION
Fluorosequencing enables researchers to compare proteins in the same sample – and across experiments – without need for external calibrants. Unlike other technologies, fluorosequencing quantifies peptides and proteins by counting molecular observations.

CHARACTERIZE HIGHLY HETEROGENEOUS SAMPLES
Our technology can discriminate peptides and proteins across a large range of heterogeneity and abundances (up to 10^6 range). Identify peptides existing in extremely diverse backgrounds, such as antigens on tumor surfaces or a low abundant phosphorylation event on a protein.
Key Concept
With help from a reference database, only a few amino acids are needed to uniquely identify a protein in a proteome.
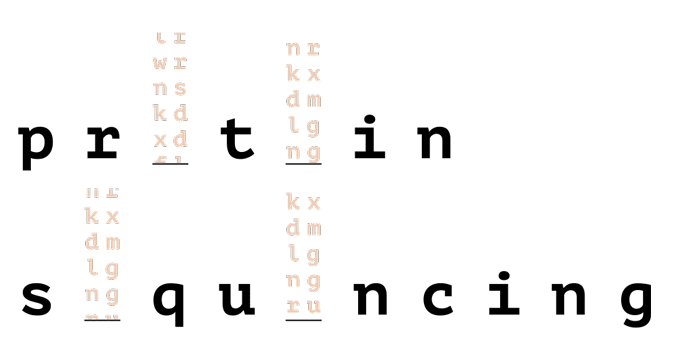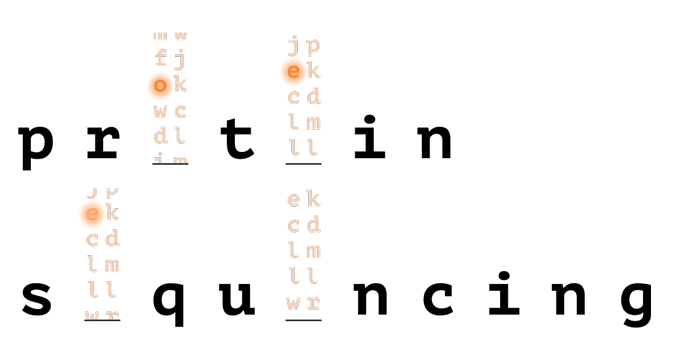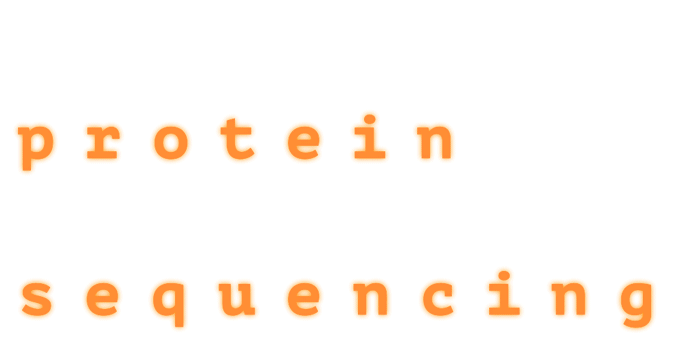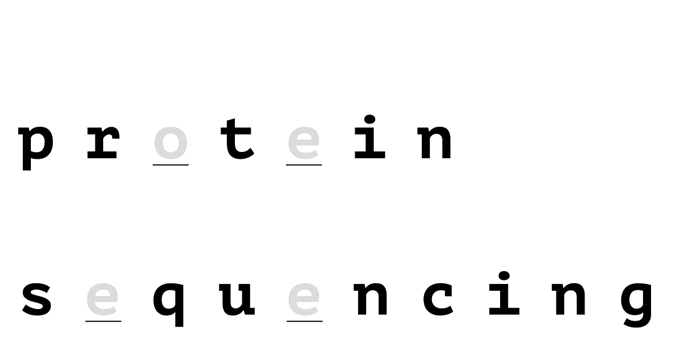
How it Works
EXAMPLE: CATALOGING ALL THE PROTEINS OF A SINGLE CELL

1.0
The protein sample is digested, creating hundreds of millions or billions of peptides.
2.0
Select amino acids are Fluorescently labeled.

3.0
The peptides are immobilized.

4.0
Image capture establishes baseline fluorescence.

5.0
The sequence is determined by the intensity of the fluorescence.
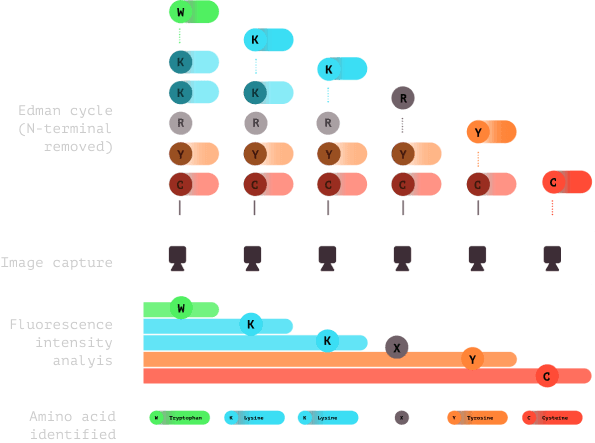
6.0
Subsequent cycles of imaging and Edman chemistry are conducted, usually 15-20 cycles. Fluorescence is analyzed and amino acids are identified to create a fluorosequence.
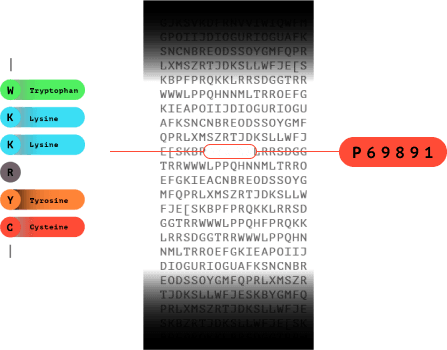
7.0
The fluorosequence is matched in a database to identify which protein the peptide came from.
FURTHER READING
NATURE BIOTECHNOLOGY
Highly Parallel Single-Molecule Identification of Proteins in Zeptomole-Scale Mixtures
WHITE PAPER
“Fluorosequencing: Concept, Features and Benefits”
APPLICATION NOTES
“Mapping the Residue Positions of a Protein’s Post- Translational Modification”
Clinical applications
and beyond
Fluorosequencing eliminates bottlenecks across biology and medicine. It will help detect disease earlier, lead to better treatments, and more.